Hydrocarbon activation has become a powerful tool in molecular science research and great progress has been made in molecular science research in recent years. In particular, weak O-coordinated carboxylic acid-assisted oxidative C-H cyclization was achieved by continuous breaking of C-H and O-H bonds of aromatic carboxylic acids (Figure 1, a). Modular assembly of five - or six-member heterocycles has made great progress under previous studies, but these reactions usually require additional use of stoichiometric chemical oxidants, such as copper salts or silver salts, to facilitate reoxidation of the catalyst.
Instead, oxidation chemical conversion is allowed through C-H bond activation, enabling reoxidation of key catalysts and avoiding the use of expensive or toxic chemical oxidants. For example, C-H cyclization via electrooxidation catalyzed by ruthenium, rhodium, and iridium (Figure 1, b). However, these catalytic processes are greatly affected by inadequate product selectivity.
Lutz Ackermann's group first reported the activation of hydrocarbons by weak O coordination of benzoic acid catalyzed by osmium (II). The advantages of osmium catalysis were highlighted through detailed reaction analysis of similar transition metal catalytic systems.
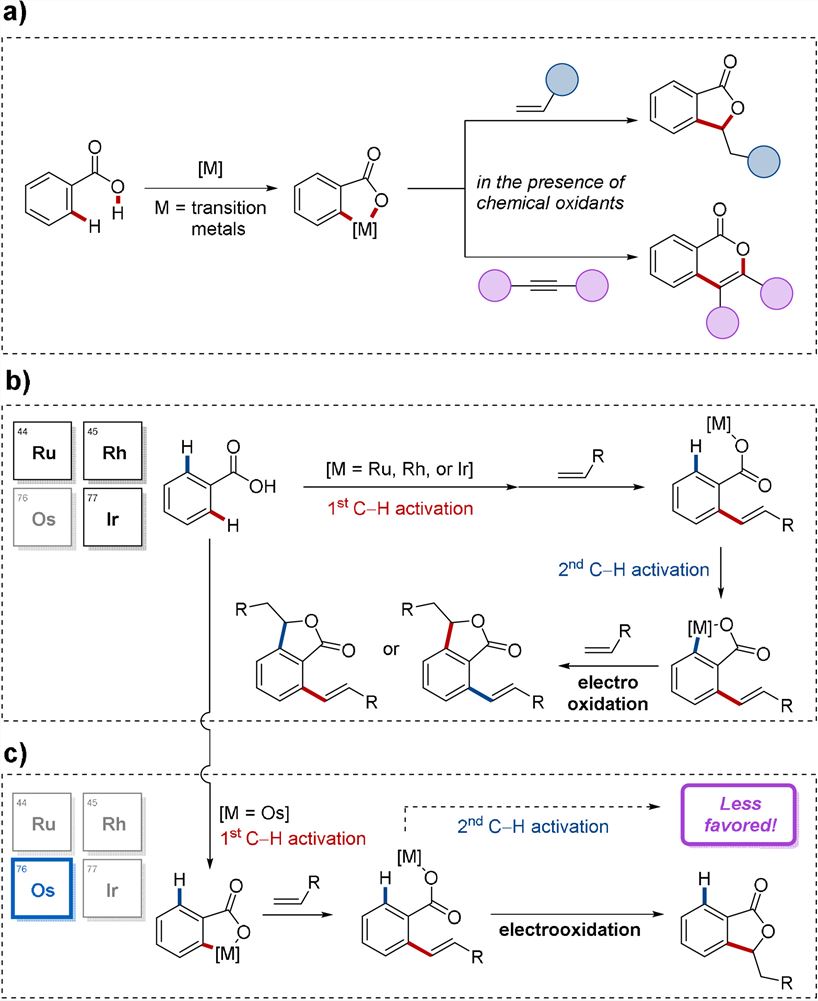 Figure 1. Transition-metal-catalyzed C-H annulations. a) General C-H annulations of benzoic acids. b) Mode of C-H activations by ruthenium, rhodium, and iridium catalysts. c) Chemoselective osmaelectrocatalyzed C-H activation.
Figure 1. Transition-metal-catalyzed C-H annulations. a) General C-H annulations of benzoic acids. b) Mode of C-H activations by ruthenium, rhodium, and iridium catalysts. c) Chemoselective osmaelectrocatalyzed C-H activation.
Osmium complexes are rarely used in organometallic catalysis as compared with their counterparts of iron and ruthenium, probably due to their lower kinetic activity. As a result, the electrooxidation of osmium catalyzed hydrocarbon activation process is difficult to control (Figure 1, c).
They hypothesized that metal centers could achieve selective control of chemical reactions by repelling steric hindrance interactions. Osmium complexes have unique physicochemical characteristics compared to other metals, so they have constructed a novel osmium (II) catalyzed electrooxidized hydrocarbon ring using weak O-oriented groups, giving the osmium complexes unique regional and chemical selectivity. According to their hypothesis, they set up feasible reaction conditions for C-H cyclization of o-toluene I with olefin II in undivided cells (Figure 2).
![Initial observation of osmaelectrocatalyzed C-H annulation. Reaction conditions: I (0.2 mmol), II (0.6 mmol), [OsCl2(p-cymene)]2 (5.0 mol %), KOAc (2.0 equiv), NaI (2.0 equiv), HFIP (2.0 mL), H2O (2.0 mL).](/upload/image/A-Strategy-for-Site-and-Chemoselective-C-H-Alkenylation-through-Osmaelectrooxidative-Catalysis-picture-2.jpg) Figure 2: Initial observation of osmaelectrocatalyzed C-H annulation. Reaction conditions: I (0.2 mmol), II (0.6 mmol), [OsCl2(p-cymene)]2 (5.0 mol %), KOAc (2.0 equiv), NaI (2.0 equiv), HFIP (2.0 mL), H2O (2.0 mL).
Figure 2: Initial observation of osmaelectrocatalyzed C-H annulation. Reaction conditions: I (0.2 mmol), II (0.6 mmol), [OsCl2(p-cymene)]2 (5.0 mol %), KOAc (2.0 equiv), NaI (2.0 equiv), HFIP (2.0 mL), H2O (2.0 mL).
The experimental results show that [OsCl2(P-Cymene)]2 combined with a Graphite felt (GF) Anode and a Platinum plate is a feasible cathode catalyst, and 29% C-H cyclization product 1 can be obtained. On this basis, they explored the influence of additives on the conductivity and reaction performance in the reaction (Figure 3).
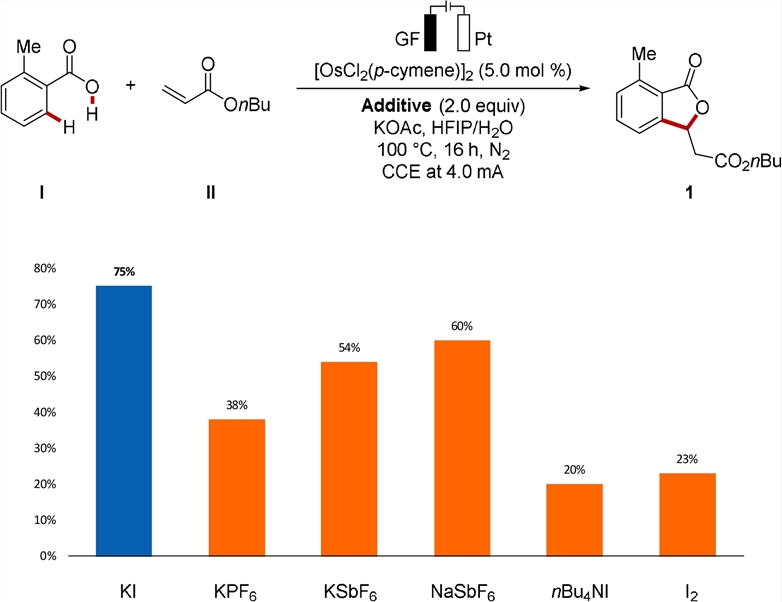 Figure 3: Examination of various additives.
Figure 3: Examination of various additives.
They added various additives, such as alkali metal base salts, organic salts and iodine, to the osmotic electrocatalytic hydrocarbon ring. Through osmotic electrocatalysis, non-toxic and inexpensive potassium iodide increased the yield of cyclated product 1 to 75%.
In addition, they conducted a set of experiments in the presence of air or common chemical oxidants such as AgOAc, Cu(OAc)2, Mn(OAc)3, PhI(OAc)2, or K2S2O8. It shows that electricity not only plays a key role in sustainable oxidation, but also provides the best and unique efficiency for C-H cyclization of osmium catalyzed oxidation (Figure 4), highlighting the great advantages of electrochemical methods.
 Figure 4: Reaction comparison between electricity and representative chemical oxidants.
Figure 4: Reaction comparison between electricity and representative chemical oxidants.
On this basis, they conducted a study on its stability (Figure 5). To this end, they performed a series of metallaelectrocatalyzed reactions with benzoic acid III possessing two accessible ortho-C-H bonds(Figure 5a). Interestingly, osmaelectrocatalysis showed unique chemoselectivity (35.5:1), while rhodium,iridium, and ruthenium catalysis gave significantly lower selectivities (5.2:1, 7.4:1, and 6.3:1 respectively). This aspect was also displayed in H/D scrambling experiments in the presence of an isotopically labeled solvent mixture (Figure 5b). With the present osmium electrocatalysis regime, deuterations occurred at the C6 position in a highly selective fashion. In sharp contrast, comparable C2 and C6 deuterations were observed in the reaction with the other Group 8 metal, ruthenium. These findings hence represent a novel tool for selective hydrogen isotopic exchange.
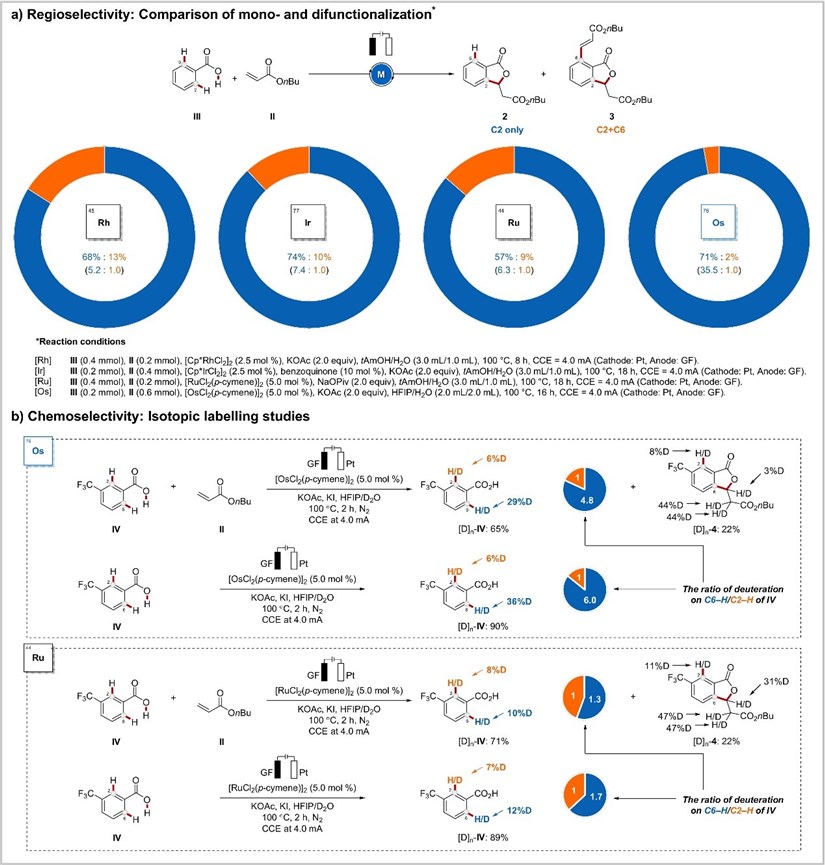 Figure 5: Selectivity of electrochemical osmium catalysis.
Figure 5: Selectivity of electrochemical osmium catalysis.
With the present osmium electrocatalysis regime, deuterations occurred at the C6 position in a highly selective fashion. In sharp contrast, comparable C2 and C6 deuterations were observed in the reaction with the other Group 8 metal, ruthenium. These findings hence represent a novel tool for selective hydrogen isotopic exchange. With these observations in mind, they set about to study the working mode of the electrooxidative osmium-catalyzed C-H annulation (Figure 6). To this end, novel key intermediates, Os-I and Os-II, were selectively prepared, which were found to afford the desired products 5 and 6, respectively, in both stoichiometric and catalytic reactions (Figure 6a). Also, in operando NMR studies revealed the consumption of Os-I and alkyne VI with concomitant formation of the new osmium(0) sandwich complex Os-II, while an induction period was not detected (Figure 6b).
Interestingly, the seven-membered intermediate formed by migratory insertion of alkyne IV was hardly observed, which presumably suggests the facile formation of the osmium(0) complex Os-II after the insertion step. Furthermore, the successive formation and consumption of key species were also clearly demonstrated by high-resolution electrospray ionization mass-spectrometric (HR-ESI-MS) analysis (Figure 6c). Additionally, cyclovoltammetric analysis of Os-II showed that the oxidation event occurred at E=0.58 V vs. Fc/Fc+, representing a slightly lower value than for the Ru0 to RuII oxidation potential (Figure 6d).
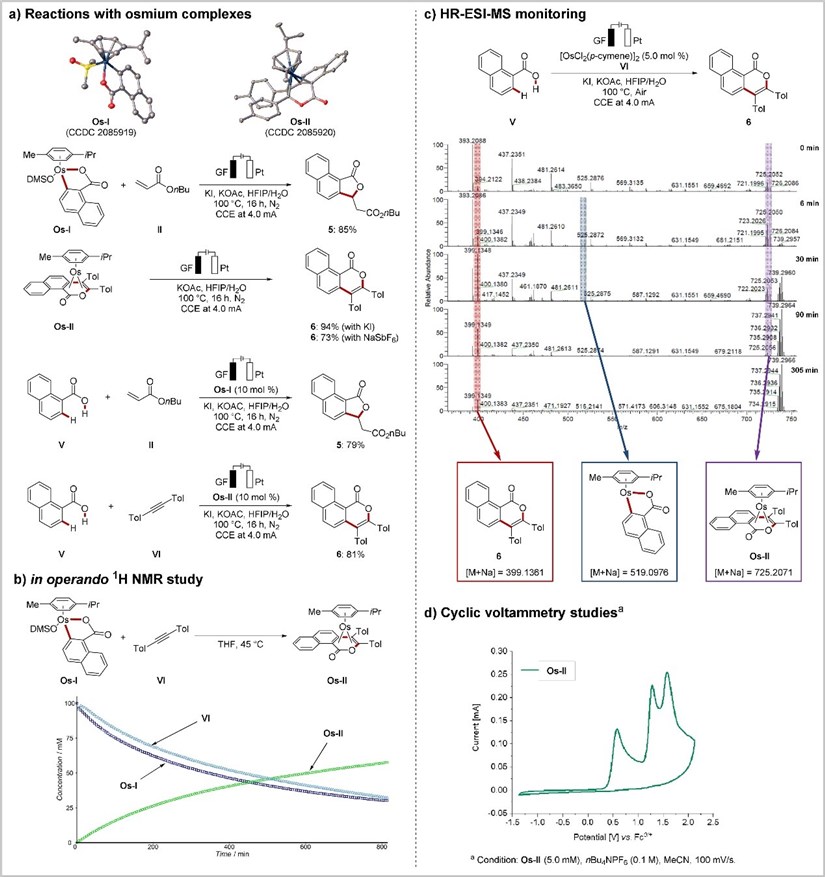 Figure 6: Mechanistic understanding of osmaelectrocatalysis.
Figure 6: Mechanistic understanding of osmaelectrocatalysis.
 Figure 7: Detailed kinetic studies and proposed mechanism.
Figure 7: Detailed kinetic studies and proposed mechanism.
Next, the performed detailed kinetic studies (Figure 7). First, intermolecular competition experiments with differently substituted benzoic acids were carried out (Figure 7a).
The competition experiments showed that electron-donating groups in benzoic acid VII gave a higher reactivity while an alkyne VI with an electron-donating group afforded less product formation. Recent studies have proposed a base-assisted internal electrophilic substitution-type(BIES) mechanism to be operative in such a scenario. Additionally, a Hammett correlation was found for benzoic acids with different substituents in the meta-position to the C-H bond to be cleaved (Figure 7b).
A linear correlation with a negative 1 value was found, indicating that an electrophilic mechanism is likely operative in the CH bond cleavage step. Importantly, we observed a considerable dependence of the conversion upon an alteration of the current, being suggestive of a turnover-limiting electron transfer step (Figure 7c).
Subsequently, an intermolecular competition reaction and a pair of parallel reactions were performed with o-toluic acid I and an isotopically labeled compound [D]7-I to determine the kinetic isotope effect (Figure 7d).
In both cases, a negligible KIE of 1.1 and 1.2 was observed, respectively, suggesting a facile C-H scission. On the basis of our mechanistic findings, a plausible catalytic cycle for the osmaelectrocatalyzed C-H activation is depicted in Figure 7e.
The mechanistic rationale commences with a facile C-H bond cleavage, which constructs osmacycle B. Thereafter, migratory insertion of the alkene or alkyne occurs, which enables the formation of intermediate D. Next, reductive elimination-in the case of a [4+1] annulation, a b-hydride elimination step is additionally involved-delivers osmium(0) sandwich complex E. Finally, the key anodic oxidation-via a proposed redox-mediator event when iodide is present- regenerates the catalytically active complex A, while liberating the desired product.
With a detailed mechanistic analysis, they eventually explored the range of viable substrates for electrochemically osmium catalyzed [4+1] and [4+2]C H/O H cyclization of benzoic acid with olefin 2 and alkyne 4(Figure 8).
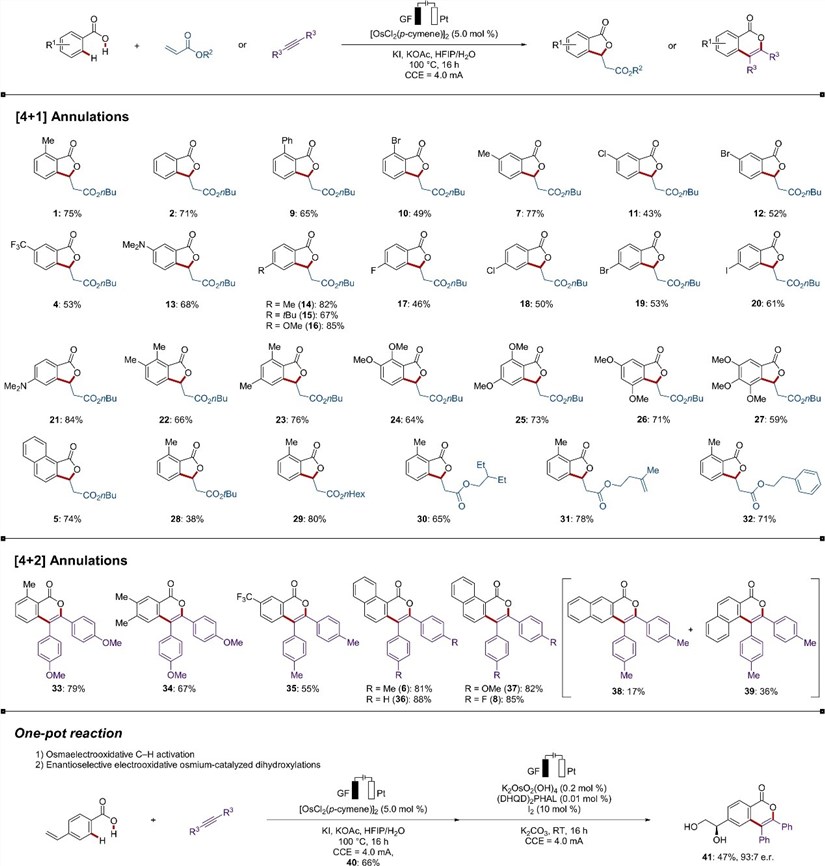 Figure 8: Versatility of the electrochemical osmium-catalyzed C-H annulation.
Figure 8: Versatility of the electrochemical osmium-catalyzed C-H annulation.
More information: Isaac, C.; et al. A Strategy for Site- and Chemoselective C-H Alkenylation through Osmaelectrooxidative Catalysis. Angew. Chem. Int. Ed., 2021, 60(2):27005-27012.
BOC Sciences explores site- and chemoselective C–H alkenylation using advanced electrooxidative catalysis methods. Our expertise ensures precise, efficient, and scalable synthetic solutions for diverse compound development.
Submit your inquiry to request a custom solution.
If you have any questions or encounter issues on this page, please don't hesitate to reach out. Our support team is ready to assist you.







