The functionalization of inactivated C- H bonds has always been an important process in organic synthesis, in which hydrogen atom transfer (HAT) has been a key initiating step for a wide range of important processes. Although chemically distinct C-H bonds can be selectively activated by the HAT processes, the regioselective activation of chemically similar C-H bonds is still difficult to achieve. For example, the Minisci reaction involving the addition of a radical to heteroarenes is a useful tool for building molecular complexity, but this reaction often requires the use of stoichiometric amount of peroxides and suffers from poor regiocontrol.
Recently, Lambert’s group at the Columbia University has developed a highly regioselective C-H functionalization method of ethers under electrophotocatalysis, published on the Journal of the American Chemical Society. The use of exogenous oxidants is avoided by the combination of electricity and photochemistry.
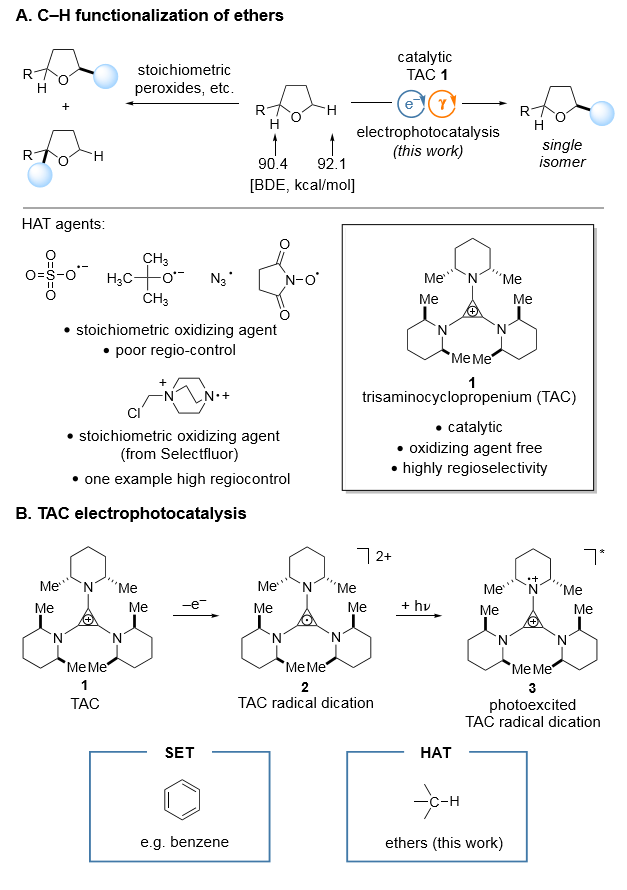 Figure 1. Electrophotocatalysis with a trisaminocyclopropenium radical dication (a) HAT process. (b) SET process.
Figure 1. Electrophotocatalysis with a trisaminocyclopropenium radical dication (a) HAT process. (b) SET process.
In the article, the researchers first studied the electrophotocatalytic coupling of THF (4) and 4-bromoisoquinoline (5)(Table 1) . In the presence of LiClO4, acetic acid, CF3CO2H and 5 mol% TAC (1), Minisci product 6 was obtained by treating the substrate with a 1.5V constant voltage and a white light compact fluorescent light (CFL) (yield 42%). Through the control experiment, the author found that light, electricity, catalyst and CF3CO2H are necessary for the reaction. By increasing the catalyst loading (8 mol%) and prolonging the reaction time (36 h), product (6) yield was increased (82%). However, when the constant voltage was increased (3.0 V), the by-products were increased, while the lower voltage did not affect the reaction. To demonstrate the preparative potential of this method, they conducted a large-scale reaction (8 mmol), which obtained 1.6g of product. Finally, the above optimization conditions were applied to a non-symmetric ether, 2-methyltetrahydrofuran (7) (entry 10), obtained a single regioisomer.
Table 1. Optimization studies

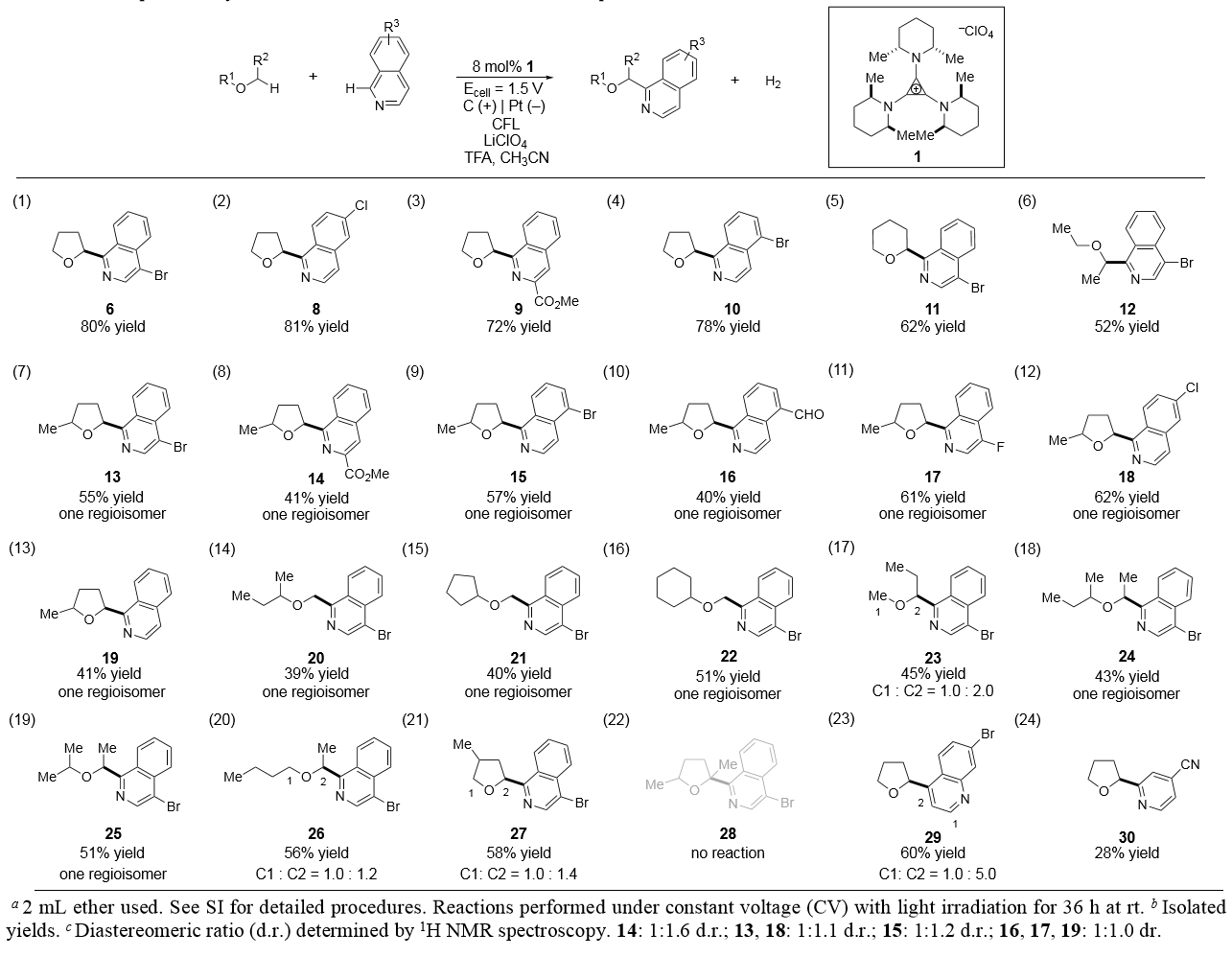
With these optimized conditions, the scope of this process was further explored. In addition to 4-bromoisoquinoline, the corresponding products were obtained by using other isoquinoline partners in moderate to good yields, and the corresponding coupling products were also obtained by other cyclic and acyclic ethers like tetrahydropyran and diethyl in moderate yields.
The key issue of regioselectivity (Table 2). 2-methyltetrahydrofuran, isoquinoline and other acyclic ethers having high regioselectivity was also studied. Compared with the tertiary C - H bonds, the primary C - H bonds were more regioselective, and only the primary C–H functionalized products were obtained. However, in the competition between primary and secondary C–H bonds, the reaction mainly took place at the secondary position, which shows excellent regioselectivity of tertiary versus secondary C-H bonds. However, for the substrates with only slightly different steric environments, essentially no regioselectivity was observed. In addition, no reaction was observed for 2,5-dimethyltetrahydrofuran, which only has tertiary a-C-H bonds. For other azole partners, the reaction with a quinoline partner was more efficient, but the efficiency of the reaction with pyridine substrate was low. Under these conditions, other species such as pyrimidine, quinoxaline, indole and so on did not react.
Table 2. Electrophotocatalytic C–H functionalization of ethers with isoquinolines and other azoles.a,b,c
Other radical acceptors were also investigated to further explore this electrophotocatalytic process (Table-3). Results showed that vinyl sulfones such as phenyl vinyl sulfone, 1,1bis(phenylsulfonyl)ethylene, acrylate and propargyl ester can obtain the corresponding products in moderate yield.
Table 3. Electrophotocatalytic C–H functionalization of ethers with alkenes and alkyne.a

In addition to the above-mentioned C-C bond coupling reactions, the electrophotocatalytic system can also recognize the formation of C - N bond (Table 4). Under the above conditions, when acetic acid is replaced by trifluoroacetic acid and the cell potential is adjusted to 2.0 V, 4-formylpyrazole and halogenated purine can react smoothly with tetrahydrofuran, tetrahydropyran or 2-methyltetrahydrofuran.
Table 4. Electrophotocatalytic C–H functionalization of ethers with azoles.a

Finally, the authors proposed the mechanism of the electrophotocatalytic reaction (Figure 2A). “The TAC cation 1 undergoes electrochemical oxidization (EOX = 1.26 V vs. SCE) to generate radical dication 2. Photoexcitation then leads to intermediate 3 bearing aminyl radical cation character. Hydrogen atom transfer from the ether substrate 7 to 3 generates the corresponding radical 46 along with the protonated TAC dication 47.”; Finally, “46 can react with isoquinoline 5 to produce intermediate radical 48, which is followed by a second oxidation (likely via 3) and loss of proton to furnish the product 12. Meanwhile, deprotonation of dication 47 regenerates the TAC catalyst 1 to close the catalytic cycle.” C-C coupling was carried out via ether radical intermediates, but the azole couplings were mechanistically ambiguous. “While they might also proceed via somophilic addition of the azole 50 to the same type of radical intermediate 49 followed by oxidation (Figure 2B, path a), it is conceivable that oxidation to the corresponding oxocarbenium ion 52 followed by nucleophile addition of the azole 50 is the operative pathway (path b).” To prove the path b is reasonable the researchers found that “the use of benzyl alcohol (53) led to acetal 54 (Figure 2C), the formation of which must have proceeded via oxocarbenium ion 52. Thus, it appears that this electrophotocatalytic strategy enables the functionalization of ethers via both one and two electron pathways using closely related conditions.”
 Figure 2. A. Mechanistic rationale for electrophotocatalytic Minisci reaction. B. Alternative pathways for azole coupling. C. Formation of acetal 54.
Figure 2. A. Mechanistic rationale for electrophotocatalytic Minisci reaction. B. Alternative pathways for azole coupling. C. Formation of acetal 54.
More information: He Huang et al. (2020). Electrophotocatalytic C–H Functionalization of Ethers with High Regioselectivity. J Am Chem Soc., 142(4):1698-1703. DOI: 10.1021/jacs.9b11472
BOC Sciences applies electrophotocatalysis for highly regioselective C–H functionalization of ethers. Our approach delivers efficient, selective, and scalable methods for complex synthesis challenges.
Submit your inquiry to request a custom solution.
If you have any questions or encounter issues on this page, please don't hesitate to reach out. Our support team is ready to assist you.







